The Emergence of the Queens-Elmhurst Hot Spot in Late March
In Part 1 of this continuing series, we offered evidence that by the first week of March 2020, SARS-CoV-2 was rapidly spreading via community transmission throughout all five boroughs of New York City. We hypothesized that the city’s massive subway system was uniquely capable of propagating the virus so widely in such a short time span. In this second part, we follow the movement of the virus beyond the first week of March, and pursue the subway hypothesis.
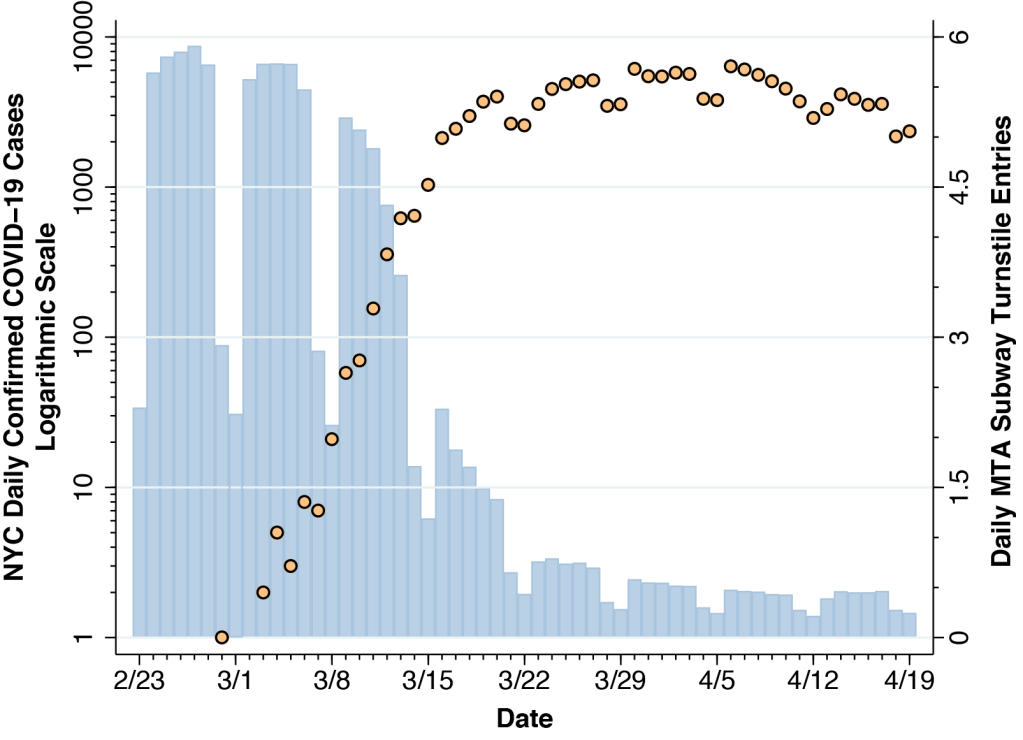
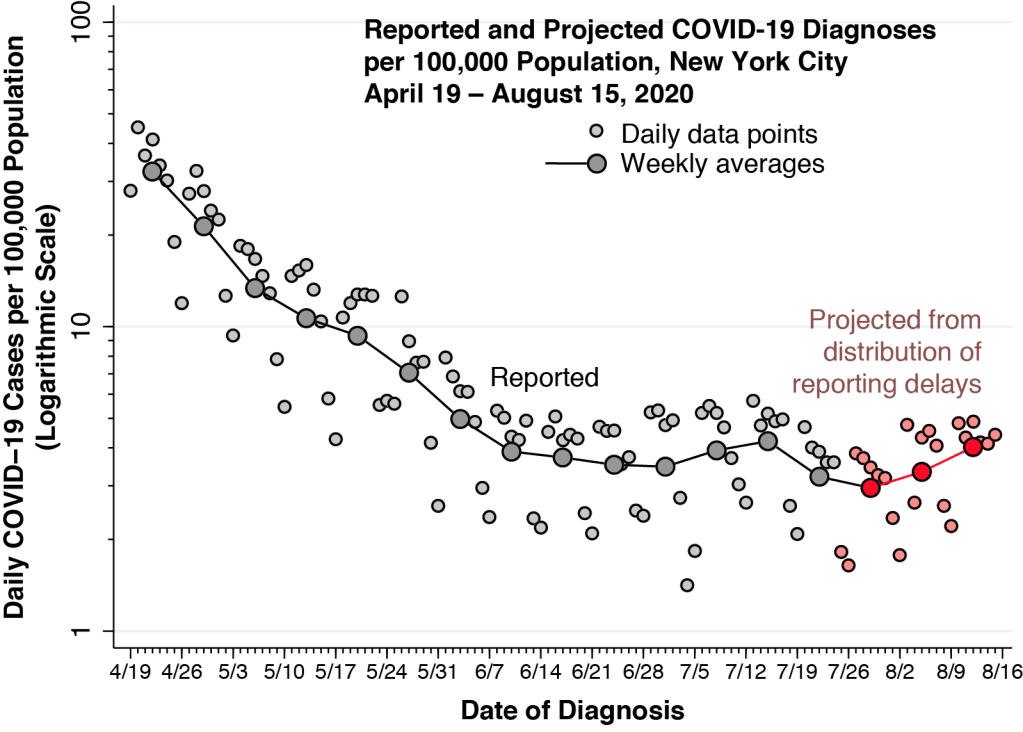
Figure 1 superimposes two time series. The mango-colored data points show the numbers of daily confirmed COVID-19 cases reported by the New York City Department of Health and Mental Hygiene. The case counts are measured on a logarithmic scale at the left. The sky-blue bars show the daily volume of trips on the city’s subway system, computed from the Metropolitan Transit Authority (MTA) turnstile data. The counts of turnstile entries are measured on a linear scale on the right-hand axis. While the computation of total subway usage entails some big-data programming, the patterns in the figure are consistent with other estimates.
The interpretation of the confirmed case counts at the end of February and during the first week of March is hampered by the restrictive testing criteria initially issued on February 28 by the U.S. Centers for Disease Control (CDC). From March 8 onward, however, once the CDC liberalized its testing criteria, we can see the rapid growth in daily confirmed cases, reaching 644 on Saturday March 14 and 1,037 by Sunday March 15.
New York’s “numbers are spiking because our testing capacity is going up,” said Gov. Andrew Cuomo in a March 14 press briefing. In fact, the upswing in cases seen in the first two weeks was entirely consistent with a massive epidemic, and not simply an artifact of increased testing. That Saturday marked the first coronavirus-related death in New York City, an 82-year-old woman admitted to Brooklyn’s Wyckoff Heights Medical Center on March 3. Given the estimated mean incubation period of 5 days, the patient was likely infected on or around February 27. With an estimated infection fatality rate of about 1 percent, that one death would imply a hundred coronavirus cases in the city by the end of February alone. If anything, the testing-confirmed case counts in the first week of March significantly understated the actual epidemic growth curve.
Subway Volume and COVID-19 Cases
During the week from March 8–15, Figure 1 above shows that subway volume was already declining from its prior average of 5.6 million turnstile entries per weekday during January and February. By Friday, March 13, volume was down to 3.6 million rides. That was two days before Mayor DeBlasio announced that he would sign an executive order closing entertainment venues and limiting restaurants to take-out and delivery, and it was four days before the order went into effect. About a week after subway volume began to decline, daily COVID-19 case counts began to deviate from their exponential trend. As subway rides fell to less than one-quarter of their regular volume in the third week of March, the epidemic curve flattened out.
The coincidence of the two data series poses a question: Does the precipitous drop in subway ridership – which began in the second week of March before the mayor ordered a shutdown – have any causal relationship to the subsequent flattening of the epidemic curve in New York City? This question is distinct from the possible role of the city’s public transportation system in the initial rapid propagation of SARS-CoV-2 throughout its five boroughs in late February and early March.
Manhattan Versus Queens

Figure 2 shows the breakdown of daily confirmed COVID-19 cases in two of the city’s boroughs: Manhattan and Queens, once again derived from data posted by the New York City department of health. During the week starting March 8, the case counts from both boroughs follow an exponential path on the graphic’s logarithmic scale, with an estimated doubling time of 1.1 days. (The estimate was based upon Poisson regression. The 95% confidence interval is shown in parentheses.)
Based on a generation time of 5.5 days, the estimated slope of 0.63/day implies a basic reproductive number of 
By the third week in March, Figure 2 shows, the two incidence curves begin to diverge significantly. By the last full week of the month, weekday reported cases in Manhattan were down to about 600, while weekday reported cases in Queens exceeded 1,500. The graphic raises yet another question: While both boroughs experienced a flattening of the epidemic curve during the last two weeks in March, why did new cases in Queens continue at more than double the rate in Manhattan?
The Queens-Elmhurst Hot Spot

Figure 3 is the earliest COVID-19 incidence map that can be constructed from publicly available data issued by the New York City health department. For each zip code, the graphic shows the cumulative number of cases per 10,000 population through March 31, 2020. While there are high-incidence zip codes in Manhattan, Brooklyn and the Bronx, there is a notable cluster of six zip codes in the Elmhurst area in Queens, especially zip codes 11367, 11368, 11369, 11370, 11372 and 11373. Even as the epidemic further expanded during April, this cluster of zip codes can be seen at the center of other incidence maps and in our own animated map. By contrast, with the exception of zip code 10018 in mid-town west near Times Square, Manhattan shows no foci of increased COVID-19 incidence.
By the end of March, it had become abundantly clear that the Elmurst area of Queens had become a coronavirus hot spot. “Queens is not the most populous borough, and it is far from the most densely populated,” wrote Clodagh McGowan for Spectrum NY1. “Experts say the borough’s demographics might play a role in why the virus has spread so quickly through Queens,” continued McGowan. “The borough is home to many city employees providing essential services – workers, immigrants and low-income service workers.”
McGowan went on to quote Dr. Sandra Albrecht, an assistant professor of Epidemiology at the Mailman School of Public Health at Columbia University: “These are folks who cannot stay at home. And even as the cases soar in the city, they have to continue taking the subway, they have to continue going to work.”
Let’s test Dr. Abrecht’s hypothesis.
The Flushing Local (Number 7) Line

Figure 4 displays the 22 stations of the Flushing Local (Number 7) line overlaid on a section of the larger March 31 zip code map shown above. We used the geographical coordinates reported by the MTA to locate each of the 22 stations on the map. The five key stations within the Queens-Elmhurst hot spot, indicated in yellow from west to east, are: 74th St – Broadway, 82nd St – Jackson Hts, 90th St – Elmhurst Av, Junction Blvd, and 103rd St – Corona Plaza. The stations within Manhattan, indicated in sky blue from west to east, are: 34th St – Hudson Yards, Times Sq – 42nd St, 5th Ave – Bryant Pk, and Grand Central – 42nd St. The remaining stations within the borough of Queens are indicated in pink.
(We excluded the 69th – Fisk Av station, located just to the west of 74th St – Broadway, because it was on the other side of I-278. We also excluded the Mets – Willets Pt station because it was in Flushing Meadows Park. It is arguable that the 111 St station, with its proximity to the Louis Armstrong House on 107th Street, should be included as part of the Corona neighborhood. Addition of that station did not change the results reported below. Nor did the exclusion of certain stations subject to temporary closures for construction and repairs.)
When Did the Hot Spot Emerge?
The maps of Figures 3 and 4 above do not by themselves tell us exactly when the cluster of zip codes in Queens emerged as a high-incidence area. However, the coincidence of the Manhattan and Queens case counts during the second week in March, shown in Figure 2, at least suggest that the hot spot did not emerge until the third week of March.
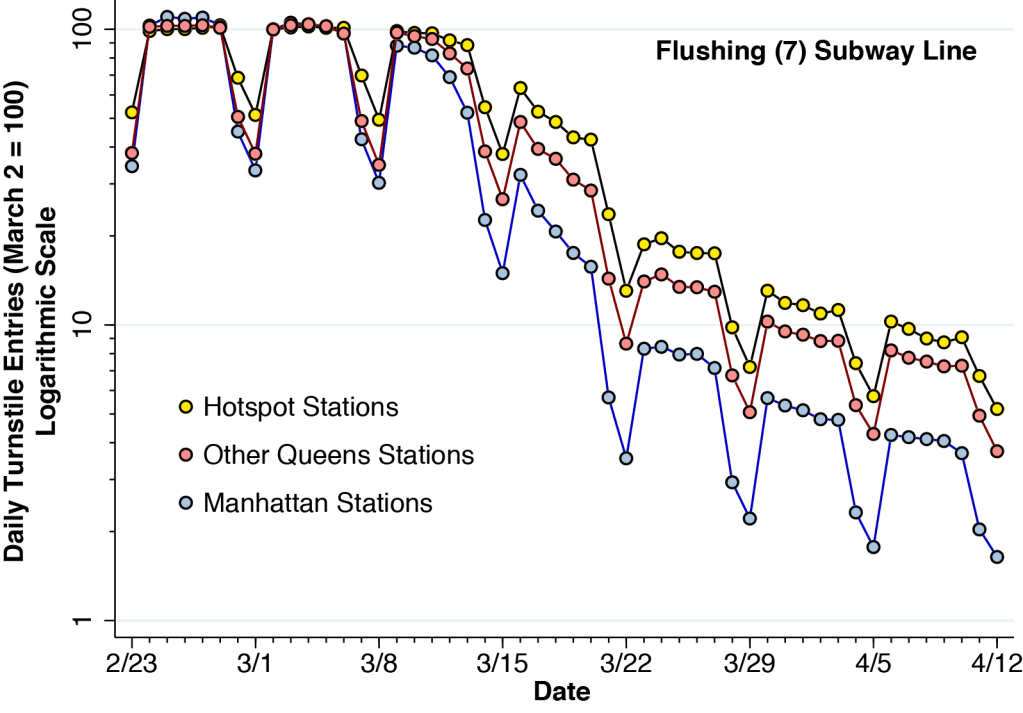
Figure 5 shows the relative numbers of daily turnstile entries for each of the three zones of the Flushing (7) line identified in Figure 4. The daily turnstile entries, likewise derived from the MTA turnstile data, were normalized so that the volume on Sunday, March 2 was equal to 100 for each zone. To show the proportional changes in volume, the vertical axis is rendered on a logarithmic scale.
Figure 5 indicates that the decline in ridership volume at the hot spot stations was slower and less extensive than in the other two zones. By Wednesday, March 18, hot spot station volume was still at 49 percent of its baseline level, while Manhattan volume was at only 21 percent of its baseline level. This odds ratio of more than 2-to-1 was maintained during the following weeks. These data support the conclusion that the more sustained subway use at the hot spot stations along the Flushing (7) line contributed at least in part to the emergence of the high-incidence of coronavirus infections in the Queens-Elmhurst area later in the month.
Manhattan Shut Down, But Queens Did Not.

Figure 6 tracks the movements of smart phone device holders who originated in one of the six zip codes in the Queens-Elmhurst hot zone. The data, which covered movements from February 23 through April 12, 2020, were derived from Safe Group Social Distancing Metrics. We previously relied on Safe Graph data in COVID-19, Bar Crowding, and the Wisconsin Supreme Court, San Antonio Conondrum, and TETRIS For Tulsa.
The Safe Graph Social Distancing Metrics data base gives the census block groups of the origin and destination of each movement resulting in a stop of at least one minute. We confined our analysis to those movements from the census block group of origin of the device to the census block group of one of three destination stations along the Flushing Local (7) line. These destination stations included: the 82 St – Jackson Heights station within the hot spot, the Queensboro Plaza station in the commercial district of Queens (located in zip code 11101), and the Times Square 42d St station located in Manhattan. (We excluded those devices whose origin was within the same census block group as the Jackson Heights station.)
The data in Figure 6, while subject to sampling error, confirm that residents of the hot spot zip codes in Queens-Elmhurst began to reduce their trips to the Times Square 42 station after March 15 and nearly stopped going there altogether by after March 22. By contrast, trips to the local Jackson Heights station and the Queensboro station declined to a much lesser extent. The data are consistent with the hypothesis that jobs in Manhattan for residents of the hot spot zip codes in Queens-Elmhurst were severely curtailed after March 15. By contrast, some residents of the Queens-Elmhurst hot spot continued to take the Flushing Local (7) line to work within the burough of Queens.

Figure 7 draws on the same Safe Graph data base to plot the movements of all Manhattan residents to the census block group for the Times Square – 42nd St subway station. The data show a near complete shut-down of traffic. In contrast to devices originating in the Queens-Elmhurst hot spot shown in Figure 6, the reduction began earlier..
What We Have and Haven’t Learned
Confirmed cases of COVID-19 in both Manhattan and Queens were already doubling every 1.1 days by the second week of March, 2020. The estimated basic reproduction number of 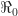
By the third week of March, the counts of new cases in different boroughs began to diverge, with less dense Queens reporting more than twice as many daily cases as Manhattan. By the end of March, a cluster of high-incidence zip codes in the Queens-Elmhurst area had been identified. While daily rider volume at the Manhattan end of the Flushing Local (Number 7) subway line dropped early and precipitously, turnstile entries into the stations within the high-incidence zip codes (the hot zone) declined more slowly and to a significantly lesser extent.
Data tracking movements of smart phone devices confirmed that residents of the hot zone virtually stopped going to work in Manhattan, but continued to take the Flushing Local to work in the commercial center of Queens. Comparable tracking of devices of Manhattan residents showed a near shutdown of trips to Times Square – 42nd St station.
Our study of the early days of the SARS-CoV-2 epidemic has actually addressed two distinct but related hypotheses. In Part 1, we inquired whether New York City’s subway system could have served as the vehicle for the massive, rapid propagation of the virus throughout the city’s five boroughs during the beginning of March. Here, we inquire whether the large-scale evacuation of the subway system was a factor in the subsequent attenuation of the epidemic during the second half of March. A corollary to the second hypothesis is that delayed, incomplete emptying of the subways in certain areas of the city contributed to the subsequent development of hot spots.
For the most part, the evidence examined in this article does not address the first hypothesis. What’s more, it addresses the second hypothesis only in connection with the Queens-Elmhurst hotspot.
With a generation time of only 5 or 6 days between when the infector contracts the virus and when the infectee does, it does not take long before secondary spread overwhelms the patterns initially established during the early phase of the epidemic. That’s one of the reasons it’s so important to study what happened in the very beginning.
But there are other important reasons to focus so sharply on the early days. This pandemic is far from over. New waves can emerge at any moment. We need to learn everything we can about the origins of the first wave if we’re to have any reasonable chance of blocking the next wave. And even if we escape this epidemic with a vaccine or a viral mutation, it should be fairly obvious from the recent cascade of MERS, SARS, H1N1 2009 and Ebola that another one is surely on its way.
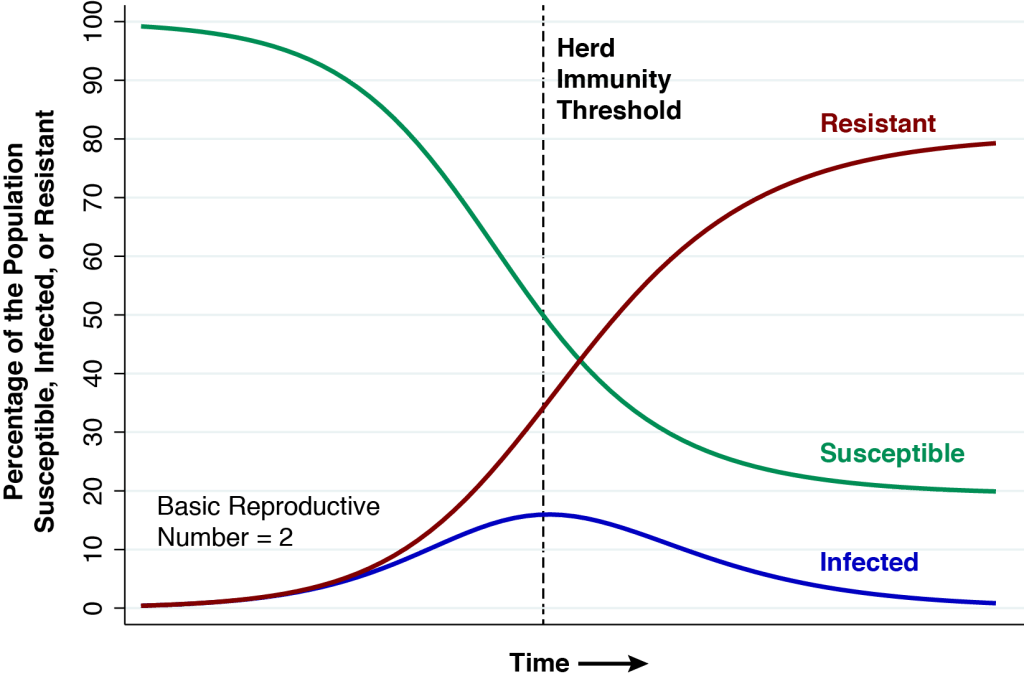
 .
. 50% = 0.25% will have died. In a population of 300 million, that’s 750,000 deaths. The point of this article is that, in fact, 80 percent will eventually be infected, so that 0.5%
50% = 0.25% will have died. In a population of 300 million, that’s 750,000 deaths. The point of this article is that, in fact, 80 percent will eventually be infected, so that 0.5%  .
.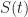 denote the proportion of the population that is susceptible to the disease at time
denote the proportion of the population that is susceptible to the disease at time 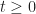 . Let
. Let  denote the proportion infected, and let
denote the proportion infected, and let 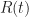 denote the proportion resistant. We assume a closed population, that is,
denote the proportion resistant. We assume a closed population, that is,
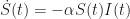
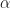 is a positive constant parameter. Here, we use the dot notation
is a positive constant parameter. Here, we use the dot notation  for the first derivative.
for the first derivative.
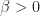 is also a constant parameter. Upon becoming infected, each individual thus remains infected for a mean time period equal to
is also a constant parameter. Upon becoming infected, each individual thus remains infected for a mean time period equal to  . Given the constraint of a closed population, the corresponding differential equation for the number of infected individuals is therefore
. Given the constraint of a closed population, the corresponding differential equation for the number of infected individuals is therefore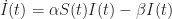
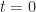 assuming everyone is naïve to the infectious agent, that is,
assuming everyone is naïve to the infectious agent, that is, 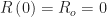 . The epidemic is initially seeded by
. The epidemic is initially seeded by 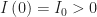 infected individuals imported from outside. The initial number of susceptible individuals is
infected individuals imported from outside. The initial number of susceptible individuals is 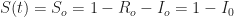 . If the initial number of infected individuals is small, then
. If the initial number of infected individuals is small, then 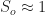 .
. , our epidemic will eventually dissipate and there will be no remaining infected individuals, that is,
, our epidemic will eventually dissipate and there will be no remaining infected individuals, that is,  . At that point, some fraction of susceptible individuals will still not have been infected. We write
. At that point, some fraction of susceptible individuals will still not have been infected. We write 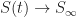 and
and  for the limiting numbers of susceptible and resistant individuals, where
for the limiting numbers of susceptible and resistant individuals, where  and
and  .
. and
and  , where
, where  . The resulting differential equation has the closed-form solution
. The resulting differential equation has the closed-form solution
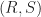 plane. At time
plane. At time 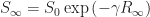 . Since
. Since 
 . In what follows, we use the fact that
. In what follows, we use the fact that 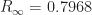 when
when  and
and  of infected individuals, that is,
of infected individuals, that is,  . We can rewrite this equation as
. We can rewrite this equation as 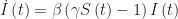 . The expression
. The expression
 gives the average number of new infections generated by a single infected individual. We let
gives the average number of new infections generated by a single infected individual. We let  denote the basic reproductive number at the start of the epidemic.
denote the basic reproductive number at the start of the epidemic. . When the reproductive number is less than 1, we’re past the herd immunity threshold and the growth rate of the infected population is negative, that is,
. When the reproductive number is less than 1, we’re past the herd immunity threshold and the growth rate of the infected population is negative, that is,  .
.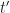 at which the epidemic reaches the herd immunity threshold. We know that
at which the epidemic reaches the herd immunity threshold. We know that 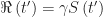 and
and  . We also have
. We also have  . So, the proportion of susceptible individuals at the herd immunity threshold equals
. So, the proportion of susceptible individuals at the herd immunity threshold equals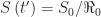
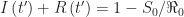 .
. , we have
, we have  . We already know that
. We already know that  , so
, so 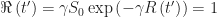 . Since
. Since 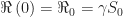 , we have
, we have  . This gives us the relation between the basic reproductive number
. This gives us the relation between the basic reproductive number  and the number of resistant individuals at herd immunity
and the number of resistant individuals at herd immunity  :
:
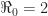 and the proportion of resistant individuals at the herd immunity threshold is
and the proportion of resistant individuals at the herd immunity threshold is 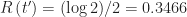 .
. and
and  and in the long run as
and in the long run as